Sickle cell disease primarily affects people of African, Mediterranean, Middle Eastern, and Asian Indian ancestry. There is also a growing segment in the Latino-American population particularly those of Caribbean, Central American, and South American ancestry. In the United States one out of every 400 births has this disease. The most common type of sickle cell disease is sickle cell anemia. In our companion article “Understanding Sickle Cell Disease” we cover a lot of material about the cause and symptoms of this disease. The following paragraph is a very brief overview.
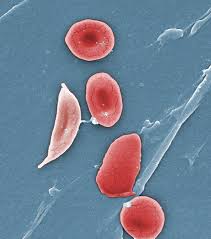
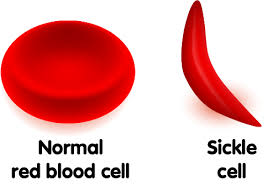
All types of sickle cell disease are caused by a genetic change in the hemoglobin portion of the red blood cell. Hemoglobin is the oxygen-carrying protein inside the red blood cell. Normal red blood cells are oval and flexible. Red blood cells in sickle cell disease have a tendency to reshape themselves into rod-like structures that resemble the curved blade of a sickle; thus, the term sickle cell. Sickle cells have a shorter life span than normal red blood cells. This results in chronic anemia and reduced oxygen to the tissues of the body. In addition, sickle cells are less flexible than normal red blood cells. This presents a problem since they can become trapped in the small blood vessels preventing blood flow to the body’s tissues. This compromise in the delivery of oxygen to the tissues results in pain and potential damage to the associated tissues and organs.
Recent Research Highlights the Importance of Nitric Oxide!
Researchers at Duke University and Howard Hughes Medical Institute recently discovered one of the keys to the cause of pain in sickle cell disease. Their findings were reported in the January 31, 2005. Proceedings of the National Academy of Sciences. Their research showed that when normal red blood cells move through the arteries they release a signaling molecule that tells the arterial walls to expand. The signaling molecule is nitric oxide. Nitric oxide causes the smooth muscle in the blood vessel wall to relax which opens up the vessel to allow the cells to pass through.
For those with sickle cell anemia, when the red blood cells are distorted into the sickle cell shape, the researchers at Duke University discovered that the walls of the arteries don’t expand. This distorted shape of the sickle cells, combined with the fact that they tend to clump together, ends up blocking blood flow through these small arteries and capillaries. The Duke researchers noted that as the blood pulses, the walls of the arteries didn’t expand like they do with normal red blood cells.
Their research also noted that the degree of nitric oxide deficiency directly correlated with symptom severity. This means that the less nitric oxide produced the greater the pain. This Duke/HHMI study found that when nitric oxide was administered to people with sickle cell anemia their symptoms were relieved. One of the conclusions from this study was that abnormal nitric oxide processing may be the real cause of sickle cell circulatory restrictions. In addition to the Duke/HHMI study, several other studies found that the administration of nitric oxide to people with sickle cell anemia relieved symptoms.
In 1998 the Nobel Prize in Medicine was award to the researchers in nitric oxide. Part of what came out of their research was that the primary pathway for creating nitric oxide in the body came from an essential amino acid called L-arginine. L-arginine is called an essential amino acid because your body cannot produce it so it must be brought into your body through your diet. L-arginine is found in foods like milk, cheese, yogurt, meat, and other proteins.
The world’s leading researcher in L-arginine is Dr. Ann de wees Allen. Dr. Allen has over 20 years of experience with L-arginine and her research has led to some breakthrough discoveries that center on the remarkable properties of L-arginine. In fact, L-arginine is considered one of the most important neutraceuticals ever developed and is referred to by scientists as the Miracle Molecule. Columbia University refers to L-arginine as the “MAGIC BULLET” for the cardiovascular system.
To be effective and safe, L-arginine must utilize a low-glycemic method. Through much research and development, Dr. Allen determined that an L-arginine molecule attached to a kiwi glycoside (from the Kiwi plant) will create the formulation that allows L-arginine to cross the proper barriers as well as allow it to be taken orally without the taste buds rejecting it. Dr. Allen formulated a product called ProArgi-9 that was granted patent #6,608,109. With this patent the FDA and FTC allowed Dr. Allen to make 15 legal claims for ProArgi-9, and legal claim #14 is “help produce Nitric Oxide.”
Two prominent researchers became aware of Dr. Allen’s work and product. Dr. Clair Francomano (former Chief of the Medical Genetics Branch at the National Institutes of Medicine, National Genome Research Institute, and Chief of Human Genetics for the Laboratory of Genetics at the National Institute on Aging) and Dr. Randall Maxey (renowned U.S. cardiologist and President, National Medical Association Research Foundation for Ethnic-Related Diseases) approached Dr. Allen regarding her patented delivery system for L-arginine and its ability to create nitric oxide safely and effectively in the human body. Because of their experience and research in genetics they were aware on an additional genetic polymorphism common to the African-American community. Many African-Americans produce too much of an enzyme called arginase. In the bloodstream, arginase destroys L-arginine so that it is no longer available for the production of nitric oxide.
With the help of Dr. Clair Francomano and Dr. Randall Maxey, Dr. Allen was able to genetically engineer a low-glycemic delivery system for L-arginine specifically designed for the African-American community. This product is call ENCODE. It is designed to help provide the body with a high level and quality of L-arginine so that proper production of nitric oxide can be achieved with the resulting health benefits that can come from nitric oxide.
14 Steps to Help Those With Sickle Cell Disease!
Understanding the past and most current research will help to chart a plan of action to help those who suffer from sickle cell disease. Please remember that these steps are for informational purposes only and is not a substitute for professional medical advice, examination, diagnosis, or treatment. Always seek the advice of a physician or other qualified healthcare provider with any questions you may have regarding this or any medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read in this article. If you think you may have a medical emergency, always call your doctor or 911 immediately.
Step 1 – Proper hydration: It is estimated that 75% of Americans have mild, chronic dehydration. This will affect blood flow so getting the proper amount of water on a daily basis is critically important. It must be pure water. Pop, coffee, diet soda, alcohol, or other doctored beverages to not count. Your body must replace 2-3 quarts of water everyday. As a rule of thumb, for every 15 lbs of weight, you need 8 ounces (1 cup) of water.
Step 2 – Stay warm: Cold reduces blood flow because capillaries constrict. If you live in a cold environment, then take extra precaution to keep your extremities, especially fingers and toes, properly protected.
Step 3 – Avoid high altitudes: Geographic locations with high elevations should be avoided due to the reduce level of oxygen in the atmosphere.
Step 4 – Exercise: Intense exercise should be avoided but mild to moderate aerobic exercise can be a benefit. Exercise improves blood flow and can produce collateral capillary beds that can aid in providing alternative pathways for blood flow when an arterial pathway becomes blocked. Before starting an exercise program, you should always consult your physician.
Step 5 – L-arginine supplementation: The body utilizes L-arginine to create nitric oxide which is necessary for the proper function of the cardiovascular system especially in the control of the elasticity of the arterial walls. Now, before you go out and start buying L-arginine supplements you need to be aware of the dark side of this amino acid. L-arginine ingested in its pure form tastes terrible and will simply go into the stomach, move into the intestinal tract and then into the gut, negating most of its benefits. There is only one way for L-arginine supplement to be safe and effective and that is in a powder form with a specific glycoside rider. By itself, L-arginine can have some serious side effects like: increasing free-radical brain damage, activation of the herpes simplex virus and reduced sperm motility. This is why L-arginine is also called the “double-edged sword.” Because L-arginine is a blind amino acid it must be attached to a rider to properly direct it to the area of the body where it will have an impact. This rider must use a low-glycemic method.
Step 6 – Organic germanium: Organic germanium was first synthesized 20 years ago. It is a trace mineral that helps support the immune system. Its many beneficial attributes include oxygen enrichment, free-radical scavenging, and heavy-metal detoxification. Organic germanium is also known to be a rejuvenator and aid in increasing stamina, endurance, energy, and heart muscle tone. Toxicological studies have documented its rapid absorption and elimination from the body. But, as a precaution, you should discuss this with both your physician and pharmacist especially if you are on any type of medication.
Step 7 – Rest: It is important to get adequate rest since stress can have a negative effect on your overall health and wellness as well as lower your immune system.
Step 8 – Support your immune system: There is so much that could be said in this area. Increasing your consumption of antioxidant rich fruits and vegetables would be an important first step. Daily consumption of at least 500 mg of vitamin C would help both the immune system and liver function.
Step 9 – Support your liver: Again, there is much that could be said in this area. Adequate amounts of green leafy vegetables with proper water intake are two good first steps to help the liver. Taking 500 mg of vitamin C will help increase the rate of synthesis of glutathione which is a major component in liver detoxification.
Step 10 – Avoid people with colds and flu: Try not to place yourself in situations that allow for infections to occur. This may require you to use a mask at times if your work or school environment is compromised with illness issues.
Step 11 – Immunizations: Stay up-to-date with the proper immunizations needed. Work with your health care provider to set up a schedule so that all the proper steps are taken to stay on top of this area.
Step 12 – Folic acid supplementation: Folic acid is a necessary component for the production of red blood cells. The recommended daily amount for adults is 400 micrograms and 600 micrograms in pregnancy. If you are thinking about taking a larger amount, then you should discuss this with both your physician and pharmacist.
Step 13 – Support groups: Because of the emotional challenges of this disease it always helps to find a support group to share frustrations, challenges, hopes, and successes.
Step 14 – Family planning: Due to the genetic nature of sickle cell disease, it is always helpful to discuss options with a family practitioner who has experience in genetic counseling.
Because of the genetic nature of sickle cell disease there is no pill to cure it. This can leave individuals and family members feeling helpless in their battle against the symptoms of this disease. Discuss these 14 steps with a qualified physician to develop a plan of action that will help to empower you to have better control over sickle cell disease.