For the African-American community, sickle cell disease is a major health issue. Sickle cell disease can also affect Mediterranean, Middle Eastern, and Asian Indian ancestry, and there is a growing segment in the Latino-American population particularly those of Caribbean, Central American, and South American ancestry. In the United States one out of every 400 births has this disease. The most common type of sickle cell disease is sickle cell anemia.
Definition and Description of Sickle Cell Disease
Sickle cell disease is a group of inherited blood disorders that center on red blood cells which can function abnormally resulting in small blood clots, chronic anemia, painful events, and potential complications associated with tissue and organ damage. These blood disorders include sickle cell anemia, Mediterranean blood disease, the sickle beta thalassemia syndromes, and hemoglobinopathies in which the sickle cell hemoglobin is in association with other abnormal hemoglobin in sufficient concentration to cause the red blood cell to sickle.
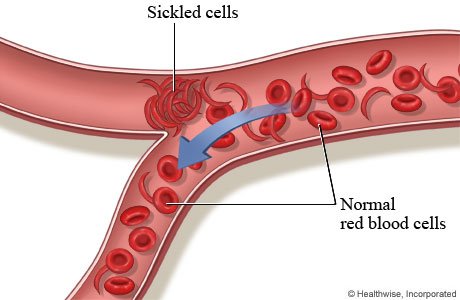
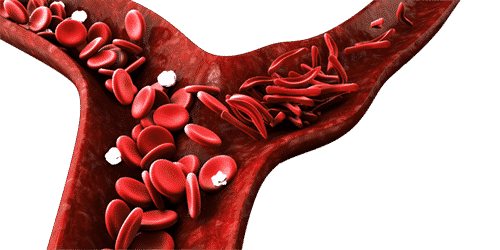
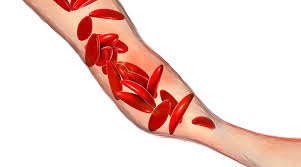
All types of sickle cell disease are caused by a genetic change in the hemoglobin portion of the red blood cell. Hemoglobin is the oxygen-carrying protein inside the red blood cell. Normal red blood cells are oval and flexible. Red blood cells in sickle cell disease have a tendency to reshape themselves into rod-like structures that resemble the curved blade of a sickle; thus, the term sickle cell. Sickle cells have a shorter life span than normal red blood cells. This results in chronic anemia and reduced oxygen to the tissues of the body. Sickle cells are less flexible and more sticky than normal red blood cells. This presents a problem since they can become trapped in the small blood vessels preventing blood flow to the body’s tissues. This compromise in the delivery of oxygen to the tissues results in pain and potential damage to the associated tissues and organs.
Carriers of the sickle cell gene are referred to as having sickle cell trait. Most of the time sickle cell trait does not cause health problems. In fact, sickle cell trait can be beneficial because it provides protection against malaria, a disease caused by blood-borne parasites transmitted through mosquito bites. It is estimated that one in 12 African-Americans has sickle cell trait.
The Cause of Sickle Cell Disease!
The hemoglobin molecule of a red blood cell is made up of three components: heme, alpha or alpha-like globin, and beta or beta-like globin. Sickle cells contain a genetic change in the beta globin component of the hemoglobin molecule. This is caused by a change in the genetic coding on chromosome 11. One small change in a single DNA nucleotide results in a different amino acid being inserted into the beta globin protein of the hemoglobin molecule resulting in the unique properties of sickle cells. For simplicity we will call this altered gene the “sickle cell gene” and the regular gene the “normal red blood cell gene.”
For most individuals, they have two copies of the “normal red blood cell gene” to produce normal beta globin resulting in typical red blood cells. Individuals with sickle cell trait have one “normal red blood cell gene” and one “sickle cell gene” so they produce both normal red blood cells and sickle cells in roughly equal proportions. Because of this they do not usually experience significant health problems as a result of having sickle cell trait. Those with sickle cell anemia have two “sickle cell genes.”
Genetics plays a significant role in both the disease, symptoms, and in family planning. If both members of a couple have sickle cell trait then there is a 25% chance in each pregnancy for the baby to inherit two sickle cell genes and the resulting child will have sickle cell anemia. Correspondingly, there is a 50% chance the baby will have sickle cell trait and a 25% chance that the baby will have the “normal red blood cell genes”. If both members of a couple have sickle cell anemia then the baby will have sickle cell anemia 100% of the time. If one member of the couple has sickle cell anemia and the other has both “normal red blood cell genes”, then the resulting child will have sickle cell trait 100% of the time. Finally, if one member of the couple has sickle cell trait and the other has both “normal red blood cell genes”, then the resulting child has a 50% chance of have normal red blood cell hemoglobin or a 50% chance of having sickle cell trait.
The Need for Oxygen!
Oxygen is necessary for life and the optimal function of all cells. Red blood cells transport oxygen from the lungs to the tissues throughout your body. It is the hemoglobin molecule that binds oxygen to itself in the lungs and then releases oxygen to the tissues for proper cell respiration. However, once the oxygen is released by the sickle cell hemoglobin it can cause the red blood cell to alter its normal oval shape into the rigid, sickle shape characteristic of sickle cells. Low oxygen can be a trigger for this change. Studies also seem to indicate that cold temperatures and dehydration can be factors in triggering this change.
Normal red blood cells can survive for approximately 120 days where as sickle cells typically last 10-12 days. This is an important factor because it leaves the bloodstream chronically short of red blood cells and hemoglobin which leads to anemia. This creates its own shortage of oxygen which could trigger a shape change in the red blood cell to the sickle shape. This rigid, sickle shape does not allow the sickle cell to fit well through small blood vessels. In addition, there are altered chemical properties that develop which increases the cells’ “stickiness”. That is why sickle cells tend to adhere to the inside surfaces of small blood vessels and other blood cells resulting in blockages in these blood vessels. These blockages prevent oxygenated blood from reaching tissue areas resulting in pain and possibly organ and tissue damage if kept without oxygen long enough.
Common symptoms include the following:
o Bloody urine, frequent urination
o Bone and/or abdominal pain, chest pain
o Delayed growth and puberty
o Excessive thirst
o Fatigue, breathlessness, rapid heart rate
o Increased susceptibility to infections, fever
o Pain which can vary from moderate to intense
o Paleness, yellow eyes and/or skin, jaundice
o Poor eyesight or blindness
o Ulcers on the lower legs usually in adolescents and adults
The severity of symptoms varies widely and cannot be predicted solely on genetic inheritance. Some with sickle cell disease develop health and life threatening problems in infancy while others only have mild symptoms throughout their lives. Others experience various degrees of health issues as they age. Certain variations of sickle cell disease tend to have less severe symptoms on average than other types of sickle cell disease.
Organs and Body Systems Affected by Sickle Cell Disease
Various organs and body systems can be effect by sickle cell disease. As you will see from this list, sickle cell disease has a wide range of effects on the body. Bottom line is that any tissue that needs oxygen and adequate blood flow can be at risk.
o Acute Chest Syndrome – Acute chest syndrome or ACS is a leading cause of death for those with sickle cell disease. It takes place in the lungs and rapid diagnosis and treatment is very important. ACS can occur at any age. It is similar to pneumonia in symptoms but distinct in its damage.
o Anemia – As we learned early, sickle cells have a life span of 10-12 days resulting in a deficiency of red blood cells in the bloodstream. It is the hemoglobin of red blood cells that carry oxygen so with this deficiency there is a reduction in oxygen to the tissues. Common symptoms of anemia include fatigue, paleness, and a shortness of breath. The heart rate will increase to try to circulate more blood to make up for the lack of oxygen to the tissues.
o Delayed Growth – Because of the short life span of sickle cells, the energy demands of the bone marrow to produce more red blood cells compete with the demands of a growing body. Children with sickle cell anemia may experience delayed growth and reach puberty at a later age. However, by early adulthood, they catch up on growth and height but may still remain below average in weight.
o Infections and the Spleen – Children under the age of three with sickle cell disease are particularly susceptible to life-threatening bacterial infections especially from Streptococcus pneumoniae. Unfortunately, 15% of these types of cases result in death. Since your spleen helps to fight bacterial infections, it is particularly vulnerable to the effects of sickle cells. It is not uncommon to see the loss of spleen function by late childhood for those with sickle cell anemia.
o Jaundice and Gallstones – Jaundice is indicated by a yellow tone in the skin and eyes due to increased levels of bilirubin which is the final product of hemoglobin degradation when red blood cells are destroyed. Bilirubin is removed from the bloodstream by the liver and elevated levels can increase the chance for gallstones.
o Joint Problems – The blood supply to the connective tissue, especially in the hip and shoulder joints, can be blocked by the sickle cells resulting in bone damage and poor healing. This complication can affect an individual’s physical abilities and result in substantial and chronic pain.
o Kidney Disease – Kidneys are particularly prone to damage from sickle cells. Adults with sickle cell disease often experience reduce functioning of the kidneys which can progress to kidney failure.
o Painful Events – This is the hallmark symptom of sickle cell disease. The frequency and duration varies tremendously from individual to individual and over an individual’s lifetime. These painful events are also the most common cause for hospitalization. The hallmark symptom results when the small blood vessels become blocked by the sickle cells preventing oxygen from reaching the tissues. Although pain can affect any area of the body, the most frequent sites are the extremities, chest, abdomen, and bones.
o Priapism – Only males have to deal with this since it is a condition characterized by a persistent and painful erection. Blood vessels become blocked by sickle cells so that blood is trapped in the tissue of the male’s organ. It is extremely painful and can result in damage to this tissue causing impotence.
o Retinopathy – The blood vessels that support the tissue at the back of the eye may be blocked by sickle cells resulting in a condition called retinopathy. Regular ophthalmology evaluations and effective treatment can help a person avoid permanent damage to their vision.
o Stroke – This is one of the most concerning complications of sickle cell disease since approximately 11% of individuals with this disease will have a recognizable stroke by the age of 20. Typically, a stroke in a person with sickle cell disease is caused by a blockage of a brain blood vessel by the sickle cells. This results in lack of oxygen to the affected area of the brain. The consequences are far ranging from undetectable effects to apparent or subtle learning disabilities to severe physical or cognitive impairment to life-threatening.
Diagnosis and Treatment
The inheritance of sickle cell disease or sickle cell trait cannot be prevented but it can be predicted so screening is recommended. If you exhibit symptoms, then contact your physician so that accurate tests can be done to determine if you carry the “sickle cell gene” and what level of risk you are at. For newborns, more than 40 states include sickle cell screening as part of the battery of blood tests. However, don’t just assume the test is done. You must always be proactive.
Hemoglobin trait screening is always a good choice for any person of a high-risk ethnic background especially if you are considering having children. If you and your partner are found to have sickle cell or other hemoglobin traits, then you might want to receive genetic counseling to better understand the risk of sickle cell disease for your offspring and various testing options available to you.
Treatment options are intended to prevent some of the symptoms and complications of sickle cell disease. These treatment options can include:
o Access to comprehensive health care
o Adequate nutrition
o Avoiding stresses and infection
o Blood transfusions
o Bone marrow transplantation
o Getting proper rest
o Good hydration
o Hydroxyurea
o Pain management
o Proper immunizations
o Supplementation with folic acid
o Support groups
o Surgery
o Use of preventative antibiotics
As with any disease condition, you want to always work with a qualified health professional to develop a course of action that best fits your individual situation.